e-oligos DNA Synthesis
DNA Synthesis
Beta-Cyanoethyl Phosphoramidite Chemistry
The synthesis of DNA from ß-cyanoethyl phosphoramidite monomers is currently the industry standard.
With this method, high coupling efficiencies are easily attained. The absence of side reactions
also confers high biological activity of the synthetic oligonucleotide.
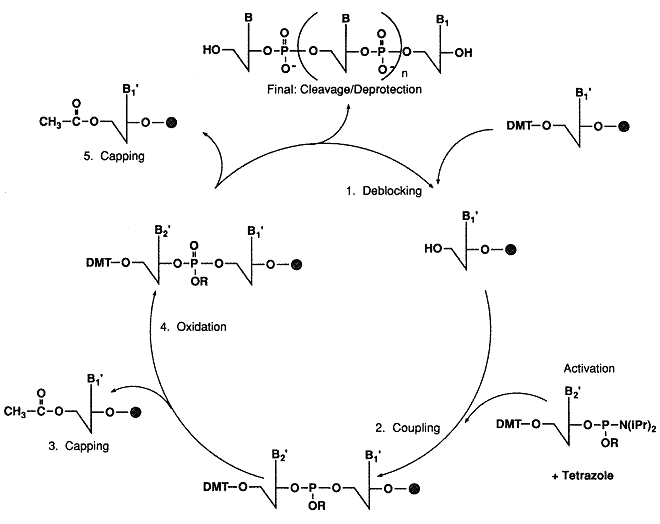
In the basic reaction cycle, a solid support, derivatized with the initial protected nucleoside,
is contained in a reaction column. Reagents and solvents are pumped through the column to effect
the addition of successive protected nucleotide monomers (phosphoramidites).
Each addition cycle includes detritylation, activation, coupling, oxidation, and capping.
Intervening wash steps remove excess reactants and by-products of reaction. After the chain
elongation is complete, the oligomer must be removed from the support and fully deprotected.
The crude product can be purified on an Oligo-Pak purification column, by gel electrophoresis, or by HPLC.
Solid Support
The synthesis of an oligonucleotide begins with the selection of the reaction column containing the initial support-bound protected nucleoside.
The reactive 5’-hydroxyl group of the support-bound nucleoside is protected with a dimethoxytrityl (DMT) group.
The 3’-hydroxyl group of the nucleoside is covalently attached, through an appropriate hydrocarbon spacer,
to the Controlled Pore Glass (CPG) support. 500A CPG is recommended for oligonucleotides up to 50 bases in length;
1000A CPG is recommended for oligonucleotides more than 50 bases in length. Following this guideline provides better coupling
efficiency by minimizing steric hindrance.
Monomers
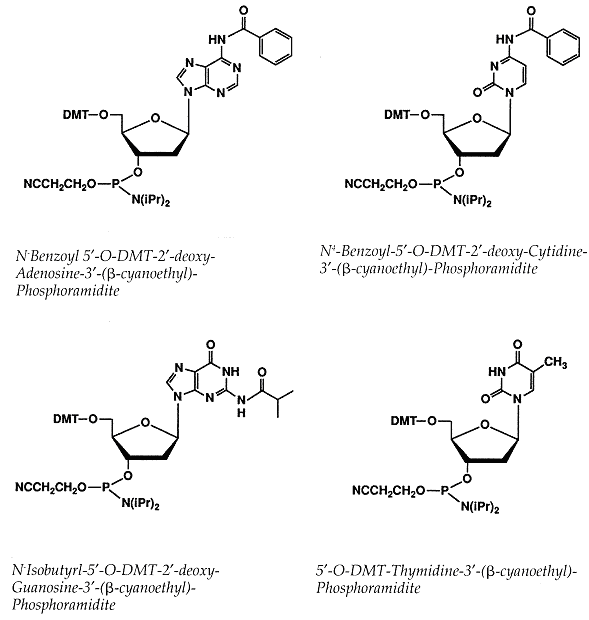
The exocyclic amines of 2’-deoxyadenosine (dA), 2’-deoxycytidine (dC) and 2’ deoxyguanosine (dG)
have to be protected during synthesis to prevent side reactions from taking place at these sites.
The dC and dA monomers are protected by benzoyl (bz) groups at the N4 and N6 positions, respectively,
while the dG is protected at the N2 position by an isobutyryl (ibu) group.
These groups are removed after synthesis of the oligonucleotide is complete, during deprotection.
The phosphoramidite monomers are also protected at the the 5’-hydroxyl position with a DMT group,
while the 3’-phosphite is modified by ß-cyanoethyl and diisopropylamine groups.
Detritylation
In the first step of the synthesis cycle, the acid labile DMT group of the support-bound monomer
is removed with a dichloroacetic acid solution (DCA). The resulting cation is orange.
The yield of the DMT cation can be estimated spectrophotometrically and be used to determine
step-wise coupling efficiency. As the DNA bases are acid-labile, the detritylation step must
only be as long as is necessary to ensure complete detritylation.
Coupling
After detritylation the next protected phosphoramidite is delivered to the reaction column.
Tetrazole is used to activate the phosphoramidite. The two reagents are mmixed just prior to delivery to the reaction column.
Tetrazole, a weak acid, protonates the tertiary nitrogen group of the phosphoramidite so that the diisopropylamine
moiety becomes a good leaving group.
The Coupling Mechanism is a nucleophylic attack by the free 5’-hydroxyl group on the 3’-phosphorous of the incoming activated monomer.
For this reason, it is important to have a totally hydroxyl-free environment in the column.
To ensure this, dry acetonitrile is used as the general solvent, and all the reagents and solvents are maintained in the anhydrous state.
Under these conditions the coupling efficiencies are very high, thereby permitting synthesis of long oligonucleotides.
Coupling
The most recently added monomer is now linked to the chain by trivalent phosphite bond.
However, phosphorus linkages are more stable when the oxidation state is pentavalent, as is the case in native DNA.
Therefore, the bond is oxidized in an iodine solution.
Capping
Since 1-2% of the free 5’-hydroxyl groups do not undergo reaction, unreacted chains
(failure sequences) must be capped to prevent further elongation in the the next cycles.
For this step, acidic anhydride and N-mehtyllimidazole are mixed to form an activated acetylating agent.
Cycling
Following the capping step, the cycle of reactions is repeated, beginning with the detritylation
step, until the chain elongation is complete.
Final Detritylation
If the oligonucleotide is to be purified by Oligo-Pak column methods or by Reverse Phase HPLC, the DMT group is
left on the 5’-OH of the oligonucleotide and is removed only after purification. If the oligonucleotide is to be purified
by gel electrophoresis or ion exchange HPLC, the oligonucleotide is detritylated at this stage.
Removal from Support and Deprotection
After the specified sequence has been assembled, the oligonucleotide must be removed (cleaved) from the support and fully deprotected prior to use.
A 90 minute room temperature treatment with ammonium hydroxide is used to cleave the oligonucleotide from the
support and to deprotect the phosphorus by ß-elimination of the cyanoethyl group. A 24 hour room
temperature treatment or an 8 hour 55’C treatment with ammonium hydroxide effectively removes the
capping groups and the benzoyl and isobutyryl groups protecting the exocyclic amines.
After cleavage/deprotection, the resulting crude mixture contains the tritylaeted product oligonucleotide,
the truncated failure sequences with free 5’-hydroxyl ends, by-products of deprotection (benzamide, isobutyramide,
acrylonitrile, and acetamide), and silicates from hydrolysis of the glass support.
Purification
The crude oligonucleotide product can be purified using reverse phase chromatography or by gel electrophoresis.
Reverse phase purification can be either processed manually using cartridges or HPLC, both methods depend on the hydrophobic trityl
group to separate the product from the failure sequences and the by-products of deprotection.
Anion exchange HPLC and gel electrophoresis separate oligonucleotides by length. Normally the product is longer than the failure sequences.
References:
Agrawal, S. (1993). Protocols for Oligonucleotides and Analogs, 20.
Agrawal S. (1994). Protocols for Oligonucleotide Conjugates, 26.
Beaucage, S. L. and Caruthers, M. H. (1981). Tetrahedron Lett., 22, 1859.
Eckstein, F. (1991). Oligonucleotides and Analogies.
Letsinger, R. L. finnan, J. L., Heavner, G. A., and
Lunsford, W. B. (1975). J. Am. Chem. Soc., 97, 3278.
McBride, L. J. and Caruthers, M. H. (1983). Tetrahedron Lett., 24, 245.
Matteucci, M. D. and Caruthers, M. H. (1980). Tetrahedron Lett. 21, 719.
|
|
